
Exploring and elaborating the novel excited state dynamical behavior of a bisflavonol system†
 *a
*a
Abstract
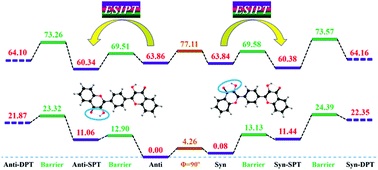
In this work, we investigate the dual hydrogen bonded 1,4-bis-(3-hydroxy-4-oxo-4H-chromen-2-yl)-benzene (bisflavonol) system in detail. Via optimizing stable structures and constructing potential energy curves, we confirm that two primary structures (i.e., anti-bisflavonol and syn-bisflavonol) can coexist in the S0 state. Calculating the reduced density gradient (RDG) versus sign(λ2)ρ and gradient isosurfaces, we confirm the formation of double hydrogen bonds for both anti-bisflavonol and syn-bisflavonol. Comparing the primary geometrical parameters involved in hydrogen bonds, we verify that the double intramolecular hydrogen bonds should be strengthened in the S1 state. In view of the photo-excitation process, we find that the charge redistributions around the hydrogen bonded moieties of both anti-bisflavonol and syn-bisflavonol facilitate the excited state intramolecular proton transfer (ESIPT) reaction. Given the reaction paths for the ESIPT process, the S0-state and S1-state potential energy surfaces (PESs) are constructed for both anti-bisflavonol and syn-bisflavonol along with two hydrogen bonds to reveal the overall excited state dynamical behavior. Searching for the transition state (TS) structure and calculating the intrinsic reaction coordinate (IRC) energetic profile, we confirm the ESIPT reaction. Combining it with Born–Oppenheimer molecular dynamics (BOMD) simulations, we study the ESIPT dynamical behaviors in detail. We present that only the single proton transfer process occurs in the S1 state in aprotic solvents, which makes up for the deficiency of previous experiments. The theoretical electronic spectra further confirm our attribution. This work not only illustrates that anti-bisflavonol and syn-bisflavonol coexisting in the S0-state can promote the respective ESIPT reaction, but also makes a new attribution to previous experiments.
 Please wait while we load your content...
Please wait while we load your content...

