
A DFT/PCM-based methodology for predicting solvolytic reactivities of organic carbonates†
 a
and
Bernard
Denegri
*a
a
and
Bernard
Denegri
*a
Abstract
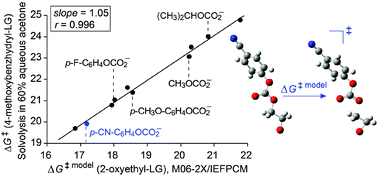
The possibility of employing quantum mechanical computations to predict solvolysis rates of benzhydryl aryl/alkyl (Ar/R) carbonates and to determine nucleofugalities of various Ar/R carbonate leaving groups in terms of Nf values is examined. Since unassisted SN1 transition states of neutral substrates cannot be optimized by using implicit solvation models, a model reaction that includes anchimerically assisted heterolysis of 2-oxyethyl Ar/R carbonates is utilized to determine the relative reactivities of both benzhydryl Ar/R carbonate substrates and Ar/R carbonate leaving groups. Very good linear correlations have been obtained between activation free energies of the model reaction, calculated by using the M06-2X method in conjunction with the IEFPCM solvation model, and activation free energies in the literature for solvolysis of the corresponding benzhydryl Ar/R carbonates in a given solvent. The slopes of close to unity demonstrate that calculated and measured relative reactivities of the Ar/R carbonate leaving groups are practically the same. Very good agreement between experiment and theory has enabled extending the nucleofugality (Nf) scale established by Mayr and co-workers (Acc. Chem. Res., 2010, 43, 1537–1549) with numerous new Ar/R carbonate leaving groups.
- This article is part of the themed collection: Mechanistic, computational & physical organic chemistry in OBC
 Please wait while we load your content...
Please wait while we load your content...

