
Thermodynamic, structural, and dynamical properties of nano-confined water using SPC/E and TIP4P models by molecular dynamics simulations†
 *a
and
*a
and
Abstract
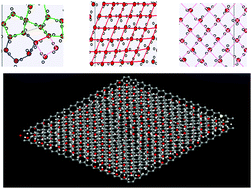
Nanoconfined water plays a significant role in nature. In recent years, many efforts have been dedicated to determine how the properties of nano-confined water change in nanopores and how these changes influence different systems, such as biological systems, and water flow in different media, such as cements and zeolites. In this work, we have simulated water molecules with different densities between two parallel graphene sheets with different distances at 300 K. Various thermodynamic, structural, and dynamical properties of the confined water molecules have been investigated using the SPC/E and TIP4P models. Our results showed that the confined systems are more stable when the pore size is between 6 and 7 Å. The energy of the confined water molecules also decreases with increasing density up to 0.6 g cc−1. Our observations showed that the confined water molecules tend to form square and rhombus structures at higher densities and tend to form pentagonal and hexagonal structures at lower densities. Pentagonal and hexagonal structures can be also more frequently observed for confined water molecules at smaller pore sizes. The most stable structure of the confined water molecules based on the SPC/E model is the rhombic structure. Our results also showed that the most stable state of the TIP4P model has fewer rhombus structures than that of the SPC/E model. Also, the TIP4P model presents greater self-diffusion values (especially at large pore sizes) than the SPC/E model. We also recognized a nanostructure phase transition from a region of high hydrogen bonding (HB) to a region of low HB by decreasing the density below around 0.6 g cc−1. The region of high HB is more ordered and contains more rhombus or square structures.
 Please wait while we load your content...
Please wait while we load your content...

