
Catalytic reduction mechanism of deoxygenation of NO via the CO-reaction pathway using nanoalloy Ag7Au6 clusters: density functional theory investigation

Abstract
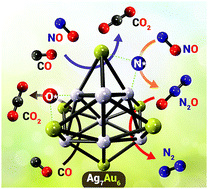
We used density functional theory calculations to investigate the catalytic potential of Ag7Au6 alloy nanoclusters for the reduction of NO by CO. The mechanism comprises two main reaction stages: reduction of NO to generate N2O followed by deoxygenation of N2O by CO to form N2 and CO2. These N2 and CO2 products desorb easily from the active Ag7Au6 site, thereby avoiding catalyst poisoning. Potential energy surfaces of the doublet- and quartet-states were systematically elucidated. No spin crossing was found during the entire reaction and our results show that the reaction preferably follows the doublet state pathway. The main reaction pathways take place at Ag7Au6 cluster facet sites, rather than at edge sites. The crucial step involves Ag7Au6-catalysed reduction of NO to generate N2O; deoxygenation of NO via the CO-reaction pathway is kinetically more favorable than that in the absence of CO. The NO reduction to generate N2O is the rate determining step with an energy barrier of 175.2 kJ mol−1. Our results reveal that this catalysed reaction is both thermodynamically and kinetically favourable. We conclude that the Ag7Au6 nanocluster has potential as a highly active catalyst for conversion of CO and NO pollutants into non-harmful products under ambient conditions.
 Please wait while we load your content...
Please wait while we load your content...

