
Molecular modelling studies of quinazolinone derivatives as MMP-13 inhibitors by QSAR, molecular docking and molecular dynamics simulations techniques†
 *a
*a
Abstract
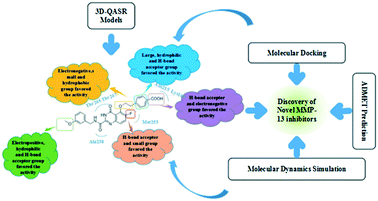
Matrix metalloproteinase-13 (MMP-13) is an attractive drug target for the treatment of osteoarthritis (OA). In this study, a series of quinazolinone derivatives as MMP-13 inhibitors were firstly systematically studied using QSAR, molecular docking and molecular dynamics (MD) simulation. The reliable CoMFA (q2 = 0.646, r2 = 0.992, Rpred2 = 0.829) and CoMSIA (q2 = 0.704, r2 = 0.992, Rpred2 = 0.839) models were constructed and verified by the Topomer CoMFA model. Results of contour maps indicated that the electrostatic, hydrophobic and H-bond acceptor fields primarily influenced the activity of MMP-13 inhibitors in the models. Several key residues (Ala238, Thr245, Thr247, Met253, Asn215 and Lys140) were identified as important factors to improve the activity and stability of the inhibitor through hydrogen bonding and electrostatic interaction. Based on these results, eight novel quinazolinones (D1–D8) were further designed. Additionally, all designed compounds showed good pharmacokinetic properties by ADMET predictions. Compounds D3 and D8 exhibited excellent predictive activity, and the 10 ns MD simulations analysis revealed that the hydrogen bonding interaction with residues (Ser250 and Gly248) was enhanced, and the small group in R2 and U-shaped conformation was of pivotal importance. These results provided strong guidance for the discovery and design of novel potential MMP-13 inhibitors.
 Please wait while we load your content...
Please wait while we load your content...