
Exploring candidate biomarkers for lung and prostate cancers using gene expression and flux variability analysis†
 ‡*ab
‡*ab
Abstract
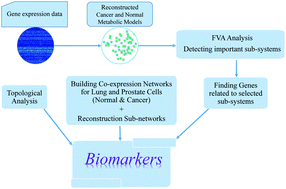
Genome-scale metabolic models have provided valuable resources for exploring changes in metabolism under normal and cancer conditions. However, metabolism itself is strongly linked to gene expression, so integration of gene expression data into metabolic models might improve the detection of genes involved in the control of tumor progression. Herein, we considered gene expression data as extra constraints to enhance the predictive powers of metabolic models. We reconstructed genome-scale metabolic models for lung and prostate, under normal and cancer conditions to detect the major genes associated with critical subsystems during tumor development. Furthermore, we utilized gene expression data in combination with an information theory-based approach to reconstruct co-expression networks of the human lung and prostate in both cohorts. Our results revealed 19 genes as candidate biomarkers for lung and prostate cancer cells. This study also revealed that the development of a complementary approach (integration of gene expression and metabolic profiles) could lead to proposing novel biomarkers and suggesting renovated cancer treatment strategies which have not been possible to detect using either of the methods alone.
 Please wait while we load your content...
Please wait while we load your content...