
Accelerating CALYPSO structure prediction by data-driven learning of a potential energy surface†
 *abc
Yanming
Ma
*abd
*abc
Yanming
Ma
*abd
Abstract
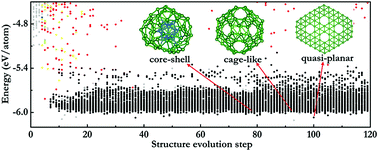
Ab initio structure prediction methods have been nowadays widely used as powerful tools for structure searches and materials discovery. However, they are generally restricted to small systems owing to the heavy computational cost of the underlying density functional theory (DFT) calculations in structure optimizations. In this work, by combining a state-of-art machine learning (ML) potential with our in-house developed CALYPSO structure prediction method, we developed two acceleration schemes for the structure prediction of large systems, in which a ML potential is pre-constructed to fully replace DFT calculations or trained in an on-the-fly manner from scratch during the structure searches. The developed schemes have been applied to medium- and large-sized boron clusters, both of which are challenging cases for either the construction of ML potentials or extensive structure searches. Experimental structures of B36 and B40 clusters can be readily reproduced, and the putative global minimum structure for the B84 cluster is proposed, where the computational cost is substantially reduced by ∼1–2 orders of magnitude if compared with full DFT-based structure searches. Our results demonstrate a viable route for structure prediction in large systems via the combination of state-of-art structure prediction methods and ML techniques.
- This article is part of the themed collection: Methods and applications of crystal structure prediction
 Please wait while we load your content...
Please wait while we load your content...

