
Modelling strategies for the covalent functionalization of 2D phosphorene†

Abstract
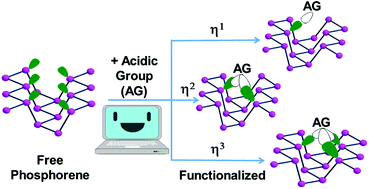
This paper is a comparative outline of the potential acid–base adducts formed by an unsaturated main group or transition metal species and P atoms of phosphorene (Pn), which derives from black phosphorus exfoliation. Various possibilities of attaining a realistic covalent functionalization of the 2D material have been examined via DFT solid state calculations. The distribution of neighbor P atoms at one side of the sheet and the reciprocal directionalities of their lone pairs must be clearly understood to foreshadow the best possible acceptor reactants. Amongst the latter, the main group BH3 or I2 species have been examined for their intrinsic acidity, which favors the periodic mono-hapto anchoring at Pn atoms. The corresponding adducts are systematically compared with other molecular P donors from a phosphine to white phosphorus, P4. Significant variations emerge from the comparison of the band gaps in the adducts and the naked phosphorene with a possible electronic interpretation being offered. Then, the Pn covalent functionalization has been analyzed in relation to unsaturated metal fragments, which, by carrying one, two or three vacant σ hybrids, may interact with a different number of adjacent P atoms. For the modelling, the concept of isolobal analogy is important for predicting the possible sets of external coligands at the metal, which may allow the anchoring at phosphorene with a variety of hapticities. Structural, electronic, spectroscopic and energy parameters underline the most relevant pros and cons of some new products at the 2D framework, which have never been experimentally characterized but appear to be reasonably stable.
 Please wait while we load your content...
Please wait while we load your content...

