
Reactions of dicobalt octacarbonyl with dinucleating and mononucleating bis(imino)pyridine ligands†

Abstract
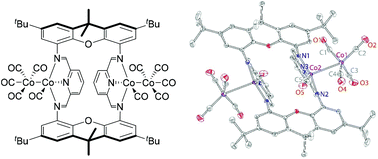
This work focuses on the application of dicobalt octacarbonyl (Co2(CO)8) as a metal precursor in the chemistry of formally low-valent cobalt with redox-active bis(imino)pyridine [NNN] ligands. The reactions of both mononucleating mesityl-substituted bis(aldimino)pyridine (L1) and dinucleating macrocyclic xanthene-bridged di(bis(aldimino)pyridine) (L2) with Co2(CO)8 were investigated. Independent of the metal-to-ligand ratio (1 : 1 or 1 : 2 ligand to Co2(CO)8), the reaction of the dinucleating ligand L2 with Co2(CO)8 produces a tetranuclear complex [Co4(L2)(CO)10] featuring two discrete [Co2[NNN](CO)5] units. In contrast, a related mononucleating bis(aldimino)pyridine ligand, L1, produces different species at different ligand to Co2(CO)8 ratios, including dinuclear [Co2(CO)5(L1)] and zwitterionic [Co(L1)2][Co(CO)4]. Interestingly, [Co4(L2)(CO)10] features metal–metal bonds, and no bridging carbonyls, whereas [Co2(CO)5(L1)] contains cobalt centers bridged by one or two carbonyl ligands. In either case, treatment with excess acetonitrile leads to disproportionation to the zwitterionic [Co[NNN](NCMe)2][Co(CO)4] units. The electronic structures of the complexes described above were studied with density functional theory. All the obtained bis(imino)pyridine complexes serve as catalysts for cyclotrimerization of methyl propiolate, albeit their reactivity is inferior compared with Co2(CO)8.
 Please wait while we load your content...
Please wait while we load your content...

