
DFT exploration of active site motifs in methane hydroxylation by Ni-ZSM-5 zeolite†

Abstract
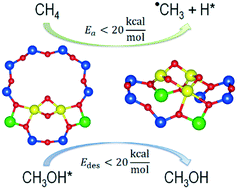
The O2-activated Ni-ZSM-5 zeolite is a promising catalyst for the selective oxidation (hydroxylation) of methane to methanol. While UV-vis spectra analyses (Shan et al. Langmuir 2014, 30, 8558–8569) have proposed bent mono(μ-oxo)dinickel [Ni2(μ-O)]2+ as the active site in Ni-ZSM-5, calculations based on density functional theory (DFT) have shown that methane activation on such an active site motif leads to a very high activation barrier, which makes it impossible for the reaction to proceed at low temperatures (<200 °C). Thus, explorations of other possible motifs of the Ni active site in ZSM-5 zeolite are indispensable. In the present study, we employed the DFT+U method to calculate methane hydroxylation on various motifs of Ni-oxo active species, including [NiO]2+, [Ni2(μ-O)]2+, [Ni2(μ-O)2]2+, and [Ni3(μ-O)3]2+, in the periodic structure of ZSM-5 zeolite. On the basis of agreement between the previously reported experimental and presently calculated activation energies, we suggest the [Ni2(μ-O)2]2+ and [Ni3(μ-O)3]2+ motifs as two possible candidates for the actual structure of active sites in Ni-ZSM-5. Different from [Cu2(μ-O)]2+-exchanged zeolites extensively studied in recent years, [Ni2(μ-O)2]2+- and [Ni3(μ-O)3]2+-ZSM-5 are predicted to activate methane and desorb the formed methanol with low activation and desorption energies, providing a new direction for low-temperature methane hydroxylation with spontaneous methanol desorption.
 Please wait while we load your content...
Please wait while we load your content...

