
The surface and catalytic chemistry of the first row transition metal phosphides in deoxygenation†
 a
and
Siris
Laursen
*a
a
and
Siris
Laursen
*a
Abstract
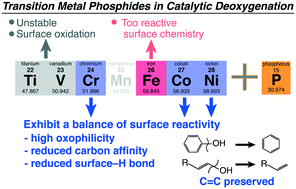
The proven utility of transition metal (TM) carbides, sulfides, and phosphides in catalytic deoxygenation reactions and their ability to preserve unsaturation or aromaticity in products has suggested the materials exhibit unique surface chemistry towards C, O, and H that is inaccessible when using reduced metal or TM + TM alloy catalysts. Herein, we present a computational surface chemistry study of the deoxygenation of phenol over most 1st row TM phosphides. Reaction mechanism analysis showed that dramatically enhanced surface reactivity towards oxygen was responsible for driving C–O cleavage. Reduced surface chemical reactivity towards carbon and limited hydrogenation activity were both beneficial in limiting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C activation in the aromatic ring and unselective overhydrogenation. The more covalent bonding within the phosphides inhibited a correlation between kinetics and thermodynamics of the C–O cleavage step due to the energetics associated with electron density transfer to or from the catalyst surface. Hydrogenation, a mostly covalent reaction step, tracked well with surface reactivity markers and d-band center of the phosphides. The less metallic electronic structure also contributed to electronically different hydrogen bonding to the surface and limited kinetics for hydrogenation that may favorably reduce unselective CC hydrogenation. Surface reaction site nature also tracked with bonding within the phosphides and their metal-to-nonmetal ratio suggesting strong electronic effects in the manipulation of the metal reaction sites and the role of nonmetal sites in the reaction mechanism.
C activation in the aromatic ring and unselective overhydrogenation. The more covalent bonding within the phosphides inhibited a correlation between kinetics and thermodynamics of the C–O cleavage step due to the energetics associated with electron density transfer to or from the catalyst surface. Hydrogenation, a mostly covalent reaction step, tracked well with surface reactivity markers and d-band center of the phosphides. The less metallic electronic structure also contributed to electronically different hydrogen bonding to the surface and limited kinetics for hydrogenation that may favorably reduce unselective CC hydrogenation. Surface reaction site nature also tracked with bonding within the phosphides and their metal-to-nonmetal ratio suggesting strong electronic effects in the manipulation of the metal reaction sites and the role of nonmetal sites in the reaction mechanism.
 Please wait while we load your content...
Please wait while we load your content...

