
Elucidating the active sites for CO2 electroreduction on ligand-protected Au25 nanoclusters†

Abstract
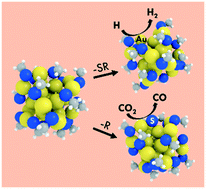
Using density functional theory (DFT) calculations, we investigated the electrochemical reduction of CO2 and the competing H2 evolution reaction on ligand-protected Au25 nanoclusters (NCs) of different charge states, Au25(SR)18q (q = −1, 0, +1). Our results showed that regardless of charge state, CO2 electroreduction over Au25(SR)18q NCs was not feasible because of the extreme endothermicity to stabilize the carboxyl (COOH) intermediate. When we accounted for the removal of a ligand (both –SR and –R) from Au25(SR)18q under electrochemical conditions, surprisingly we found that this is a thermodynamically feasible process at the experimentally applied potentials with the generated surface sites becoming active centers for electrocatalysis. In every case, the negatively charged NCs, losing a ligand from their surface during electrochemical conditions, were found to significantly stabilize the COOH intermediate, resulting in dramatically enhanced CO2 reduction. The generated sites for CO2 reduction were also found to be active for H2 evolution, which agrees with experimental observations that these two processes compete. Interestingly, we found that the removal of an –R ligand from the negatively charged NC, resulted in a catalyst that was both active and selective for CO2 reduction. This work highlights the importance of both the overall charge state and generation of catalytically active surface sites on ligand-protected NCs, while elucidating the CO2 electroreduction mechanisms. Overall, our work rationalizes a series of experimental observations and demonstrates pathways to convert a very stable and catalytically inactive NC to an active electrocatalyst.
 Please wait while we load your content...
Please wait while we load your content...

