
Elucidating metal hydride reactivity using late transition metal boryl and borane hydrides: 2c–2e terminal hydride, 3c–2e bridging hydride, and 3c–4e bridging hydride†‡
 *a
*a
Abstract
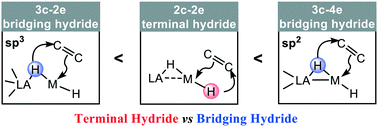
Metal hydrides play important roles in catalysis for sustainable energy, the environment, the petrochemical industry, and many important chemical processes. Despite this significance, the mystery behind metal hydride reactivity still remains. This theoretical study reveals a surprising reactivity discrepancy for different types of terminal hydrides and bridging hydrides, with Lewis acid–transition metal (LA–TM) hydride complex promoted alkene hydrogenations as model reactions, using density functional theory (DFT) studies. PBP(μ-H)CoH and DPB(μ-H)NiH complexes were chosen as representative models for the boryl type and the borane type LA–TM hydride, respectively. The bridging hydride is less reactive than the terminal hydride in the borane type complex DPB(μ-H)NiH. However, in sharp contrast, the bridging hydride is more reactive than the terminal hydride in the boryl type complex PBP(μ-H)CoH. Typical features of the electronic structure are unfolded to rationalize the origin of the reactivity discrepancy. The bridging hydride in the sp3 borane type DPB(μ-H)NiH complex forms a typical three-center two-electron (3c–2e) B–H–Ni bond. In the sp2 boryl PBP(μ-H)CoH complex, the bridging hydride forms an unusual three-center four-electron (3c–4e) B–H–Co bond. The 3c–2e bridging hydride is stabilized by two LA sites, leading to a lower nucleophilicity than that of a normal 2c–2e terminal hydride. Meanwhile the 3c–4e bridging hydride shows a stronger free-hydride character, resulting in a higher nucleophilicity than that of a 2c–2e terminal hydride. A general hydride nucleophilic trend is proposed: 3c–4e bridging hydride > 2c–2e terminal hydride > 3c–2e bridging hydride. These fundamental aspects of metal hydride reactivity should be helpful for mechanistic understanding and catalyst/material design involving metal hydride complexes.
 Please wait while we load your content...
Please wait while we load your content...

