
O-Substituted group-controlled selectivity in Rh(iii)-catalyzed coupling of benzamides with α,α-difluoromethylene alkynes: a computational mechanistic study†

Abstract
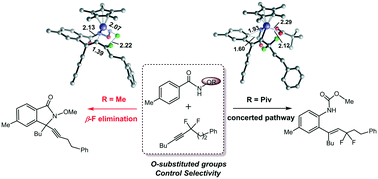
The Rh(III)-catalyzed C–H activation of benzamide derivatives with α,α-difluoromethylene alkynes was studied by DFT calculations to elucidate the selectivity controlled by different O-substituted groups (OPiv versus OMe). The reactions share a similar process that involves N–H deprotonation, C–H activation and alkyne insertion to form a 7-membered rhodacycle. Then the reaction pathway bifurcates for the two different directing groups. For OPiv, the following step involves concerted ipso attack and pivalate migration, methanol nucleophilic addition and protonation to generate hydroarylation products. For OMe, the subsequent pathway contains the first β-F elimination, intramolecular aminorhodation and the second β-F elimination to produce [4 + 1] annulation products. The N–O bond strength and the coordination of the O-substituted groups to Rh are responsible for the selectivity. The carbonyl oxygen in the OPiv group provides crucial rhodium–oxygen interaction, which essentially stabilizes the 7-membered intermediate and thus facilitates the concerted ipso attack and pivalate migration process, while the much stronger N–O bond of the OMe-containing 7-membered rhodacycle promotes the β-F elimination.
 Please wait while we load your content...
Please wait while we load your content...

