
Exploring perovskites for methane activation from first principles
 *a
*a
Abstract
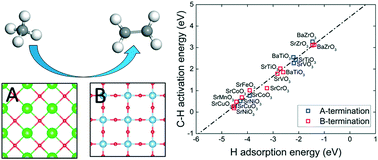
The diversity of perovskites offers many opportunities for catalysis, but an overall trend has been elusive. Using density functional theory, we studied a large set of perovskites in the ABO3 formula via descriptors of oxygen reactivity such as vacancy formation energy, hydrogen adsorption energy, and the first C–H activation energy of methane. It was found that changing the identity of B within a period increases the oxygen reactivity from the early to late transition metals, while changing A within a group has a much smaller effect on oxygen reactivity. Within the same group, B in the 3d period has the most reactive lattice oxygen compared to the 4d or 5d period. Some perovskites display large differences in reactivity for different terminations. Further examination of the second C–H bond breaking on these perovskites revealed that larger A cations and non-transition metal B cations have higher activation energies, which is conducive to the formation of coupling products instead of oxidation to CO or CO2. Balance between the first C–H bond breaking and methyl desorption suggests a just right oxygen reactivity as described by the hydrogen adsorption energy. These insights may help in designing better perovskite catalysts for methane activation.
 Please wait while we load your content...
Please wait while we load your content...

