
Ab initio electronic structure calculations of entire blue copper azurins†

Abstract
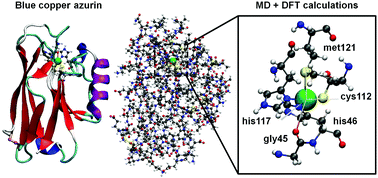
We present a theoretical study of the blue-copper azurin extracted from Pseudomonas aeruginosa and several of its single amino acid mutants. For the first time, we consider the whole structure of this kind of protein rather than limiting our analysis to the copper complex only. This is accomplished by combining fully ab initio calculations based on density functional theory with atomic-scale molecular dynamics simulations. Beyond the main features arising from the copper complex, our study reveals the role played by the peripheral parts of the proteins. In particular, we find that oxygen atoms belonging to carboxyl groups which are distributed all over the protein contribute to electronic states near the HOMO. The contribution of the outer regions to the electronic structure of azurins had so far been overlooked. Our results highlight the need to investigate them thoroughly; this is especially important in prospect of understanding complex processes such as the electronic transport through metal–metalloprotein–metal junctions.
 Please wait while we load your content...
Please wait while we load your content...

