
A theoretical study of the photodynamics of salicylidene-2-anthrylamine in acetonitrile solution†

Abstract
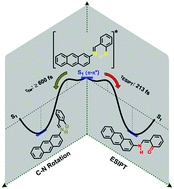
The ultrafast photoinduced processes of salicylidene-2-anthrylamine (2-AntSA) in acetonitrile solution have been investigated using DFT/TD-DFT static electronic structure calculations and excited state ab initio molecular dynamics simulations. Two different isoenergetic enol ground-state structures with suitable geometry for excited state intramolecular proton transfer (ESIPT) where chosen for the excited-state dynamics. The S1 relaxed potential energy profiles for the excited state intramolecular proton transfer and the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond isomerization reactions predict that both reactions occur over an energy barrier and that they are competitive processes in the deactivation of the Franck–Condon state. The photodynamic simulations show that the ESIPT occurs in the femtosecond time scale for both conformers (77 and 213 fs for IA and IIA, respectively) and that the speed is modulated by the ability of the conformers to evolve toward a planar conformation in the S1 state. The trajectories predict two conical intersections which provide nonradiative relaxation pathways to the S0 state. The first one is located in the twisted enol region, where the proton transfer process is unlikely, and only occurs for the conformer IIA in a time scale ≥600 fs. The second conical intersection is located in the cis-keto region, and represents an effective depopulation channel toward the trans-keto form. All our results are in remarkably good agreement with experiments.
C double bond isomerization reactions predict that both reactions occur over an energy barrier and that they are competitive processes in the deactivation of the Franck–Condon state. The photodynamic simulations show that the ESIPT occurs in the femtosecond time scale for both conformers (77 and 213 fs for IA and IIA, respectively) and that the speed is modulated by the ability of the conformers to evolve toward a planar conformation in the S1 state. The trajectories predict two conical intersections which provide nonradiative relaxation pathways to the S0 state. The first one is located in the twisted enol region, where the proton transfer process is unlikely, and only occurs for the conformer IIA in a time scale ≥600 fs. The second conical intersection is located in the cis-keto region, and represents an effective depopulation channel toward the trans-keto form. All our results are in remarkably good agreement with experiments.
 Please wait while we load your content...
Please wait while we load your content...

