
Atomistic modelling of entropy driven phase transitions between different crystal modifications in polymers: the case of poly(3-alkylthiophenes)†
 a
Tommaso
Nicolini,
‡a
Antonino
Famulari,
a
Guido
Raos,
a
Riccardo
Po
b
and
Stefano V.
Meille
*a
a
Tommaso
Nicolini,
‡a
Antonino
Famulari,
a
Guido
Raos,
a
Riccardo
Po
b
and
Stefano V.
Meille
*a
Abstract
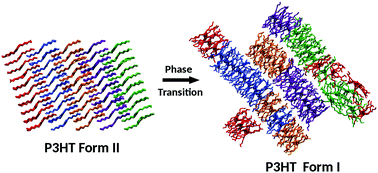
Polymorphism and related solid-state phase transitions affect the structure and morphology and hence the properties of materials, but they are not-so-well understood. Atomistic computational methods can provide molecular-level insights, but they have rarely proven successful for transitions between polymorphic forms of crystalline polymers. In this work, we report atomistic molecular dynamics (MD) simulations of poly(3-alkylthiophenes) (P3ATs), widely used organic semiconductors to explore the experimentally observed, entropy-driven transition from form II to more common form I type polymorphs, or, more precisely, to form I mesophases. The transition is followed continuously, also considering X-ray diffraction evidence, for poly(3-hexylthiophene) (P3HT) and poly(3-butylthiophene) (P3BT), evidencing three main steps: (i) loss of side chain interdigitation, (ii) partial disruption of the original stacking order and (iii) reorganization of polymer chains into new, tighter, main-chain stacks and new layers with characteristic form I periodicities, substantially larger than those in the original form II. The described approach, likely applicable to other important transitions in polymers, provides previously inaccessible insight into the structural organization and disorder features of form I structures of P3ATs, not only in their development from form II structures but also from melts or solutions.
 Please wait while we load your content...
Please wait while we load your content...

