
First principles study on 2H–1T′ transition in MoS2 with copper†

Abstract
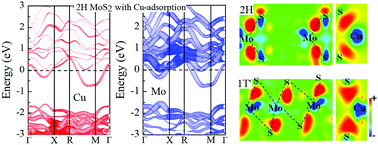
The electronic properties of MoS2 are strongly controlled by the structure, providing a route to their modulation. We report, based on first principles calculations, that the adsorption of metal atom Cu on the surface can induce the phase transition of MoS2 from the semiconducting 2H to the metallic 1T′ phase. Cu adsorption results in effective n-type doping of MoS2 by charge transfer from Cu in the case of the 1T′ phase. This is distinct from the behavior in the 2H phase, where Cu does not donate any charge, and it is also distinct from alkali metal adsorption, where charge is donated to both 2H and 1T′ MoS2. Charge donation to the 1T′ phase by Cu stabilizes it with respect to the 2H structure and importantly, it also reduces the energy barrier between the 2H and 1T′ structures. This difference reflects the higher electronegativity of Cu, which also indicates that Cu-modified MoS2 can be expected to be less chemically reactive than MoS2 with alkali metal adatoms. The main atomic mechanism of the structural transition is the gliding of S atoms on the upper surface. Finally, we report the energetics of the 2H to 1T′ transition with several other adatoms, Ag, Au, Ni, Pt and Pd, but none of them are as effective as Cu in inducing the transition.
 Please wait while we load your content...
Please wait while we load your content...

