
Ab initio molecular dynamics study of solvated electrons in methanol clusters†
 *c
*c
Abstract
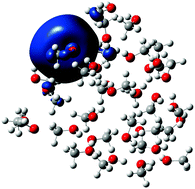
We performed a series of ab initio molecular dynamics simulations to investigate the physical properties of small methanol cluster anions, [(CH3OH)n]−, (n = 8–32). An excess electron was attached to neutral clusters that were prepared to accommodate the electron in interior cavity states or surface bound states. The computed initial binding energies of the electrons to these clusters indicate appealing similarity to the experimentally observed vertical detachment energies. The tendency of the interior state clusters parallels that of the clusters with strong electron binding in the experiments, while the simulated unrelaxed surface state anions are similar to the observed weakly bound species. This assignment is consistent with a previous identification based on hybrid quantum-classical simulations. The time evolution of the cluster anions suggests that interior state electrons slowly move to and relax on the surface, in excess electronic states that appear significantly more stable than the experimentally assigned putative surface states. Based on this result we predict the existence of relaxed surface state isomers of small methanol cluster anions. Due to the kinetic metastability of the experimentally found weakly bound species, we anticipate a serious technical challenge to prepare and identify small methanol cluster anions with relaxed surface states. These more strongly binding surface states are stabilized by dangling hydroxyl hydrogen atoms pointing to the excess electron's charge distribution. In addition, methyl hydrogens also appear to contribute to the stability of these states. During its transition to the surface, the interior excess electron maintains its initial solvent cavity. No signs of non-cavity interior states are observed in the present first principles ab initio molecular dynamics simulations.
 Please wait while we load your content...
Please wait while we load your content...

