
Core electron binding energies of adsorbates on Cu(111) from first-principles calculations†

Abstract
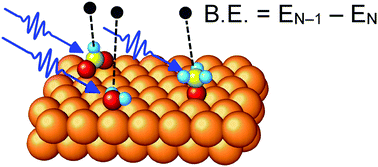
Core-level X-ray Photoelectron Spectroscopy (XPS) is often used to study the surfaces of heterogeneous copper-based catalysts, but the interpretation of measured spectra, in particular the assignment of peaks to adsorbed species, can be extremely challenging. In this study we present a computational scheme which combines the use of slab models of the surface for geometry optimization with cluster models for core electron binding energy calculation. We demonstrate that by following this modelling strategy first principles calculations can be used to guide the analysis of experimental core level spectra of complex surfaces relevant to heterogeneous catalysis. The all-electron ΔSCF method is used for the binding energy calculations. Specifically, we calculate core-level binding energy shifts for a series of adsorbates on Cu(111) and show that the resulting C1s and O1s binding energy shifts for adsorbed CO, CO2, C2H4, HCOO, CH3O, H2O, OH, and a surface oxide on Cu(111) are in good overall agreement with the experimental literature.
 Please wait while we load your content...
Please wait while we load your content...

