
Unveiling the effects of A-site substitutions on the oxygen ion migration in A2−xA′xNiO4+δ by first principles calculations†

Abstract
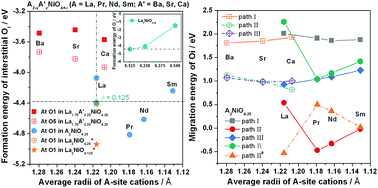
The effects of A-site substitutions on the interstitial oxygen formation energy and the migration energy in layered A2−xA′xNiO4+δ (A = selected lanthanides, A′ = Ba, Sr, Ca) are investigated by first principles calculations. The interstitial oxygen formation energy is negative, in the range of −4.81 eV to −3.45 eV, strongly supporting easiness of formation of the interstitial oxygen defects in the (A,A′)O rock salt plane. The Pr2NiO4+δ compound shows the lowest formation energy, indicating the highest amount of interstitial oxygen. Doping with alkaline earth cations (A′) increases the formation energy of the interstitial oxygen, which prefers to be located far away from the dopants. Nevertheless, Ca seems to be the best choice, due to relatively low formation energy. Calculations for the four kinds of diffusion paths allow it to be predicted that the oxygen transport in A2−xA′xNiO4+δ is governed by the interstitialcy mechanism in the ab plane, because of the significantly lower energy barriers for this mechanism. An interesting finding is achieved for A2NiO4+δ (A = Pr, Nd, Sm), for which the energy barriers for the interstitialcy transport are negative (−0.47 eV, −0.33 eV and −0.02 eV, respectively), implying that the transition state is more stable than the assumed initial state. A new structural configuration is proposed in this work, with the adjacent apical oxygen located at the adjacent interstitial site, which shows ca. 0.5 eV lower free energy than that of the initial model. This result provides a new understanding for the location of the interstitial and the adjacent apical oxygens from an energetic point of view and supports previously published experimental data. It is found that alkaline earth doping at the A-site deteriorates the interstitial oxygen diffusion in La2−xA′xNiO4.25 materials, but concerning overall transport properties, Ca seems to be a good dopant from an energetic point of view, when compared with Ba and Sr.
 Please wait while we load your content...
Please wait while we load your content...

