
How well can density functional theory and pair-density functional theory predict the correct atomic charges for dissociation and accurate dissociation energetics of ionic bonds?†

Abstract
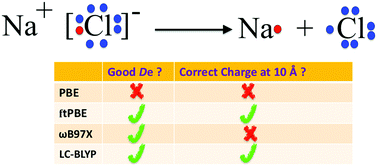
The accuracy of density functional theory (DFT) is often judged by predicted dissociation energies, but one should also consider charge densities as illustrated here for dissociation of heteronuclear diatomic molecules, including ionic bonds for which local density functionals yield erroneous results. Some hybrid density functionals with 100% exact exchange in Kohn–Sham DFT and the local functionals in multiconfiguration pair-density functional theory give relatively acurate dissociation energies for NaCl, and they correctly yield uncharged dissociated atoms.
 Please wait while we load your content...
Please wait while we load your content...

