
Assessment of electronic transitions involving intermolecular charge transfer in complexes formed by fullerenes and donor–acceptor nanohoops†

Abstract
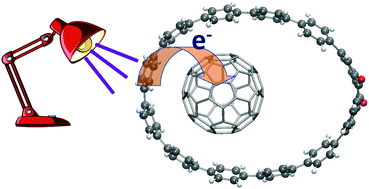
Complexes formed by fullerenes C60/C70 and substituted cycloparaphenylenes with the capability of acting as donor/acceptor pairs ([10]CPAq and [10]CPTcaq nanohoops) have been studied using density functional theory methods empirically corrected for dispersion. All nanohoops form stable complexes with fullerenes, with complexation energies amounting to around −32 kcal mol−1 with C60 and reaching between −36 and −39 kcal mol−1 in the case of C70. According to DFT calculations, the rings are too large to appropriately accommodate the fullerene, which moves from the centre of the ring to a side region (in most cases located on the side opposite the anthracene unit). In this way, the rings are deformed (the oval is stretched) in order to better accommodate the fullerene. Anyway, the orientation and position of the fullerenes inside the nanohoop have moderate influence on the stability. The preference for a given structure is the result of the combined effect of larger dispersion energies and smaller energy costs associated with the deformation of the ring. The analysis of the electronic transitions of the different complexes suggests that the presence of the anthracene unit promotes the appearance of intermolecular (nanohoop to fullerene) charge transfer transitions, even though no direct participation of the substituent has been observed.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

