
Water dissociation on K2O-pre-adsorbed transition metals: a systematic theoretical study†
 *ab
*ab
Abstract
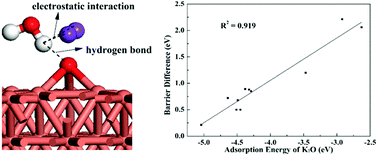
It is imperative to regulate O–H bond cleavage on metal surfaces with a pre-adsorbed K2O promoter in heterogeneous catalysis. Density functional theory (DFT) calculations have been performed to explore the adsorption and dissociation of water on K2O-pre-adsorbed transition metal surfaces (Au, Ag, Cu, Ni, Pt, Rh, Ir, Pd, Ru, Co and Fe) as compared with those on clean and K-pre-adsorbed metal surfaces. The calculation results indicate that the presence of K2O species significantly promotes water dissociation and the promoting effect depends on the adsorption strength of K2O, namely, the more strongly K2O binds to the metal surface, the less promoting effect it has on the water O–H bond cleavage. Based on geometrical and electronic analysis, the stronger promoting effect of K2O than K on water dissociation on the given metal surfaces can be attributed to stronger attractive electrostatic interactions between OH and the dissociating H of H2O at the TSs as well as between O of H2O and K of K2O at the ISs on K2O-pre-adsorbed surfaces compared with those on K-pre-covered surfaces. Moreover, the additional hydrogen bond interaction between H and Oad of K2O at the ISs on Cu/Ag/Au and Fe/Co/Ni metals would be responsible for the much greater promoting effect of K2O than K on these metal surfaces, while there is a slightly greater promoting effect of K2O on the remaining metal surfaces. From the above analysis, we expect our studies can provide profound understanding of the nature of the promoting effect of K2O on O–H bond scission.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

