
Ab initio molecular dynamics of atomic-scale surface reactions: insights into metal organic chemical vapor deposition of AlN on graphene

Abstract
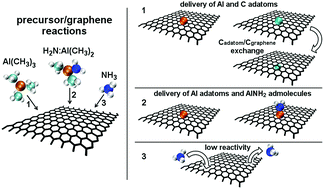
Metal organic chemical vapor deposition (MOCVD) of group III nitrides on graphene heterostructures offers new opportunities for the development of flexible optoelectronic devices and for the stabilization of conceptually-new two-dimensional materials. However, the MOCVD of group III nitrides is regulated by an intricate interplay of gas-phase and surface reactions that are beyond the resolution of experimental techniques. We use density-functional ab initio molecular dynamics (AIMD) with van der Waals corrections to identify atomistic pathways and associated electronic mechanisms driving precursor/surface reactions during metal organic vapor phase epitaxy at elevated temperatures of aluminum nitride on graphene, considered here as model case study. The results presented provide plausible interpretations of atomistic and electronic processes responsible for delivery of Al, C adatoms, and C–Al, CHx, AlNH2 admolecules on pristine graphene via precursor/surface reactions. In addition, the simulations reveal C adatom permeation across defect-free graphene, as well as exchange of C monomers with graphene carbon atoms, for which we obtain rates of ∼0.3 THz at typical experimental temperatures (1500 K), and extract activation energies Eexca = 0.28 ± 0.13 eV and attempt frequencies Aexc = 2.1 (×1.7±1) THz via Arrhenius linear regression. The results demonstrate that AIMD simulations enable understanding complex precursor/surface reaction mechanisms, and thus propose AIMD to become an indispensable routine prediction-tool toward more effective exploitation of chemical precursors and better control of MOCVD processes during synthesis of functional materials.
 Please wait while we load your content...
Please wait while we load your content...

