
The stability, electronic structure, and optical absorption of boron-nitride diamondoids predicted with first-principles calculations†

Abstract
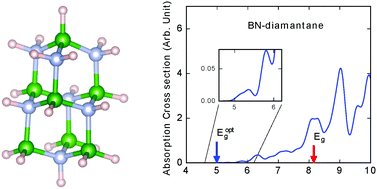
Although diamondoids are broadly studied for their fundamental properties and applications, boron-nitride-based diamondoids are scarcely explored. Here we predict the stability, electronic structure, and optical absorption spectra of six boron-nitride (BN) diamondoids with first-principles methods based on pseudopotential density functional theory and many-body perturbation methods implemented with a real-space formalism. We find that four of them are thermodynamically stable at room temperature, while B10N8H24 and B6N4H16 show thermodynamic instability in molecular dynamics simulations. With the GW approximation, we predicted the ionization energies and electron affinities of BN-diamondoids and find that the evolution of the electronic structure with size does not follow the same trend as diamondoids, owing to the unbalanced numbers of boron and nitrogen atoms. We show strong photoabsorption of BN-triamantane and BN-adamantane in the infrared and visible ranges and analyze the features of low-energy absorption by examining the characteristics of related orbitals.
 Please wait while we load your content...
Please wait while we load your content...

