
Ab initio investigation of O2 adsorption on Ca-doped LaMnO3 cathodes in solid oxide fuel cells

Abstract
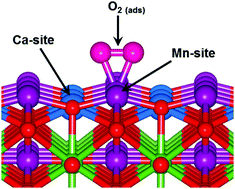
We present a Hubbard-corrected density functional theory (DFT+U) study of the adsorption and reduction reactions of oxygen on the pure and 25% Ca-doped LaMnO3 (LCM25) {100} and {110} surfaces. The effect of oxygen vacancies on the adsorption characteristics and energetics has also been investigated. Our results show that the O2 adsorption/reduction process occurs through the formation of superoxide and peroxide intermediates, with the Mn sites found to be generally more active than the La sites. The LCM25{110} surface is found to be more efficient for O2 reduction than the LCM25{100} surface due to its stronger adsorption of O2, with the superoxide and peroxide intermediates shown to be energetically more favorable at the Mn sites than at the Ca sites. Moreover, oxygen vacancy defect sites on both the {100} and {110} surfaces are shown to be more efficient for O2 reduction, as reflected in the higher adsorption energies calculated on the defective surfaces compared to the perfect surfaces. We show from Löwdin population analysis that the O2 adsorption on the pure and 25% Ca-doped LaMnO3 surfaces is characterized by charge transfer from the interacting surface species into the adsorbed oxygen πg orbital, which results in weakening of the O–O bonds and its subsequent reduction. The elongated O–O bonds were confirmed via vibrational frequency analysis.
 Please wait while we load your content...
Please wait while we load your content...

