
Modeling TADF in organic emitters requires a careful consideration of the environment and going beyond the Franck–Condon approximation†‡

Abstract
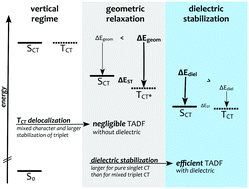
The origin of the thermally-activated delayed fluorescence (TADF) of three organic emitters is investigated, focusing on the nature of the lowest excited states, their transition properties, as well as the role of the environment. For this purpose, the algebraic-diagrammatic construction for the polarization propagator at second order of perturbation theory [ADC(2)], time-dependent density-functional theory in the Tamm–Dancoff approximation (TDA) and unrestricted Kohn–Sham DFT in combination with the maximum-overlap method (ΔDFT) are employed. The influence of the dielectric environment is rigorously included using different variants of the polarizable continuum model. The calculations reveal the lowest excited singlet and triplet states of all studied emitters to be dominated by charge-transfer (CT) character already in the most apolar environment corresponding to cyclo-hexane. The dielectric stabilization entails a drastic reduction of the singlet–triplet gaps, increasing the calculated TADF rates by several orders of magnitude. Another ingredient for accurate TADF rates is hidden in the excited-state potential-energy surfaces along the donor–acceptor twisting angle. A presence of large, shallow plateaus in apolar environments causes the transition properties to be governed by thermal fluctuations rather than the minimum-energy geometries. This leads to a large increase of the oscillator strengths, as well as a breakdown of the Franck–Condon approximation. The last ingredient is a small but significant spin–orbit coupling (SOC) between the singlet and triplet CT states, which is traced back to a delocalization of the excitation hole or excited electron of the triplet CT state. Taking into account all of these effects, a reasonable agreement with experimental TADF and fluorescence lifetimes is obtained. For this, it proves to be sufficient to consider only the lowest lying singlet and triplet excited states.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

