
Tracking the energy flow in the hydrogen exchange reaction OH + H2O → H2O + OH

Abstract
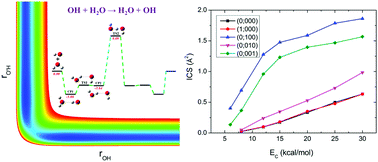
The prototypical hydrogen exchange reaction OH + H2O → H2O + OH has attracted considerable interest due to its importance in a wide range of chemically active environments. In this work, an accurate global potential energy surface (PES) for the ground electronic state was developed based on ∼44 000 ab initio points at the level of UCCSD(T)-F12a/aug-cc-pVTZ. The PES was fitted using the fundamental invariant-neural network method with a root mean squared error of 4.37 meV. The mode specific dynamics was then studied by the quasi-classical trajectory method on the PES. Furthermore, the normal mode analysis approach was employed to calculate the final vibrational state distribution of the product H2O, in which a new scheme to acquire the Cartesian coordinates and momenta of each atom in the product molecule from the trajectories was proposed. It was found that, on one hand, excitation of either the symmetric stretching mode or the asymmetric stretching mode of the reactant H2O promotes the reaction more than the translational energy, which can be rationalized by the sudden vector projection model. On the other hand, the relatively higher efficacy of exciting the symmetric stretching mode than that of the asymmetric stretching mode is caused by the prevalence of the indirect mechanism at low collision energies and the stripping mechanism at high collision energies. In addition, the initial collision energy turns ineffectively into the vibrational energy of the products H2O and OH while a fraction of the energy transforms into the rotational energy of the product H2O. Fundamental excitation of the stretching modes of H2O results in the product H2O having the highest population in the fundamental state of the asymmetric stretching mode, followed by the ground state and the fundamental state of the symmetric stretching mode.
 Please wait while we load your content...
Please wait while we load your content...

