
CO2 adsorption on gas-phase Cu4−xPtx (x = 0–4) clusters: a DFT study†

Abstract
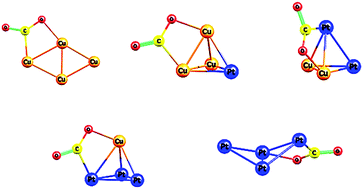
Transition and noble metal clusters have proven to be critical novel materials, potentially offering major advantages over conventional catalysts in a range of value-added catalytic processess such as carbon dioxide transformation to methanol. In this work, a systematic computational study of CO2 adsorption on gas-phase Cu4−xPtx (x = 0–4) clusters is performed. An exhaustive potential energy surface exploration is initially performed using our recent density functional theory basin-hopping global optimization implementation. Ground-state and low-lying energy isomers are identified for Cu4−xPtx clusters. Secondly, a CO2 molecule adsorption process is analyzed on the ground-state Cu4−xPtx configurations, as a function of cluster composition. Our results show that the gas-phase linear CO2 molecule is deformed upon adsorption, with its bend angle varying from about 132° to 139°. Cu4−xPtx cluster geometries remain unchanged after CO2 adsorption, with the exception of Cu3Pt1 and Pt4 clusters. For these particular cases, a structural conversion between the ground-state geometry and the corresponding first isomer configurations is found to be assisted by the CO2 adsorption. For all clusters, the energy barriers between the ground-state and first isomer structures are explored. Our calculated CO2 adsorption energies are found to be larger for Pt-rich clusters, exhibiting a volcano-type plot. The overall effect of a hybrid functional including dispersion forces is also discussed.
- This article is part of the themed collection: Celebrating recent chemical science in Mexico
 Please wait while we load your content...
Please wait while we load your content...

