
Degradation paths of manganese-based MOF materials in a model oxidative environment: a computational study†
 *ab
*ab
Abstract
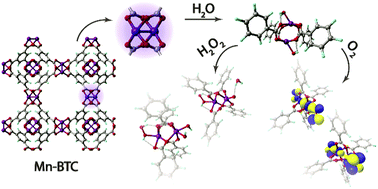
Stability is the key property of functional materials. In this work we investigate computationally the degradative potential of a model Mn-BTC (BTC = benzene-1,3,5-tricarboxylate) metal–organic framework (MOF) building block in aqueous solutions under oxidative conditions. Model density functional theory calculations have shown that the direct hydrolysis of the Mn-containing moieties is more difficult than their decomposition via oxidation-induced paths. While the interaction with H2O2 species is of non-covalent nature and requires O–O-bond breaking to initiate Mn-center oxidation, open-shell O2 species readily oxidize radical Mn-centers and form bonds of σ-, π-, or δ-symmetry with the metal. The oxidative transformations of di-Mn paddle-wheel carboxylate structure-forming units are accompanied with substantial distortions of the coordination polyhedra that, together with the increased Lewis acidity of the oxidized metal centers, facilitates the hydrolysis leading to the degradation of the structure at a larger scale. Whereas such a mechanism is expected to hamper the catalytic applications of such Mn-MOFs, the associated structural response to oxidizing and radical species can create a basis for the construction of Mn-MOF-based drug delivery systems with increased bio-compatibility.
 Please wait while we load your content...
Please wait while we load your content...

