
Solvent-dependent dual fluorescence of the push–pull system 2-diethylamino-7-nitrofluorene†

Abstract
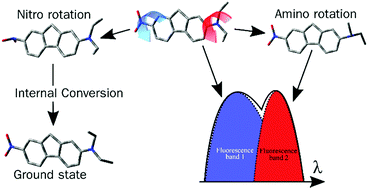
The solvent-dependent excited state behavior of the molecular push–pull system 2-diethylamino-7-nitrofluorene has been explored using femtosecond transient absorption spectroscopy in combination with density functional theory calculations. Several excited state minima have been identified computationally, all possessing significant intramolecular charge transfer character. The experimentally observed dual fluorescence is suggested to arise from a planar excited state minimum and another minimum reached by twisting of the aryl–nitrogen bond of the amino group. The majority of the excited state population, however, undergo non-radiative transitions and potential excited state deactivation pathways are assessed in the computational investigation. A third excited state conformer, characterized by twisting around the aryl–nitrogen bond of the nitro group, is reasoned to be responsible for the majority of the non-radiative decays and a crossing between the excited state and ground state is localized. Additionally, ultrafast intersystem crossing is observed in the apolar solvent cyclohexane and rationalized to occur via an El-Sayed assisted transition from one of the identified excited state minima. The solvent thus determines more than just the fluorescence lifetime and shapes the potential energy landscape, thereby dictating the available excited state pathways.
 Please wait while we load your content...
Please wait while we load your content...

