
Tunable electronic properties of partially edge-hydrogenated armchair boron–nitrogen–carbon nanoribbons†
 *bc
*bc
Abstract
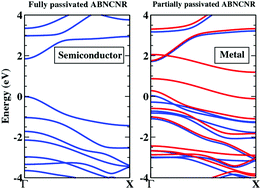
We employ a first-principles calculations based density-functional-theory (DFT) approach to study the electronic properties of partially and fully edge-hydrogenated armchair boron–nitrogen–carbon (BNC) nanoribbons (ABNCNRs), with widths between 0.85 nm to 2.3 nm. Due to the partial passivation of edges, the electrons, which do not participate in the bonding, form new energy states located near the Fermi-level. Because of these additional bands, some ABNCNRs exhibit metallic behavior, which is quite uncommon in armchair nanoribbons. Our calculations reveal that metallic behavior is observed for the following passivation patterns: (i) when the B atom from one edge and the N atom from another edge are unpassivated. (ii) when the N atoms from both the edges are unpassivated. (iii) when the C atom from one edge and the N atom from another edge are unpassivated. Furthermore, spin-polarization is also observed for certain passivation schemes, which is also quite uncommon for armchair nanoribbons. Thus, our results suggest that the ABNCNRs exhibit a wide range of electronic and magnetic properties in that the fully edge-hydrogenated ABNCNRs are direct band gap semiconductors, while the partially edge-hydrogenated ones are either semiconducting, or metallic, while simultaneously exhibiting spin polarization, based on the nature of passivation. We also find that the ribbons with larger widths are more stable as compared to the narrower ones.
 Please wait while we load your content...
Please wait while we load your content...

