
Design of donor–acceptor copolymers for organic photovoltaic materials: a computational study†

Abstract
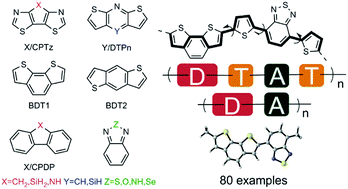
80 different push–pull type organic chromophores which possess Donor–Acceptor (D–A) and Donor–Thiophene–Acceptor–Thiophene (D–T–A–T) structures have been systematically investigated by means of density functional theory (DFT) and time-dependent DFT (TD-DFT) at the B3LYP/6-311G* level. The introduction of thiophene (T) in the chain has allowed us to monitor the effect of π-spacers. Benchmark studies on the methodology have been carried out to predict the HOMO and LUMO energies and optical band gaps of the D–A systems accurately. The HOMO and LUMO energies and transition dipoles are seen to converge for tetrameric oligomers, and the latter have been used as optimal chain length to evaluate various geometrical and optoelectronic properties such as bond length alternations, distortion energies, frontier molecular orbital energies, reorganization energies and excited-state vertical transition of the oligomers. Careful analysis of our findings has allowed us to propose potential donor–acceptor couples to be used in organic photovoltaic cells.
 Please wait while we load your content...
Please wait while we load your content...

