
Ab initio derived group additivity model for intramolecular hydrogen abstraction reactions†

Abstract
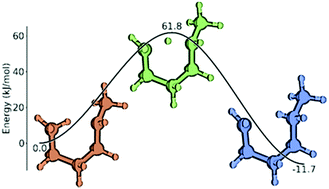
A set of group additivity values for intramolecular hydrogen abstraction reactions of alkanes, alkenes and alkynes is reported. Calculating 448 reaction rate coefficients at the CBS-QB3 level of theory for 1-2 up to 1-7 hydrogen shift reactions allowed the estimation of ΔGAV° values for 270 groups. The influence of substituents on (1) the attacking radical, (2) the attacked carbon atom, and (3) the carbon chain between the attacking and attacked reactive atom has been systematically studied. Substituents have been varied between hydrogen atoms and sp3, sp2 and sp hybridized carbon atoms. It has been assumed that substituents further away from the reactive atoms or their connecting carbon chain have negligible influences on the kinetics. This group additivity model is applicable to a wide variety of reactions in the 300–1800 K temperature range. Correlations for tunneling coefficients have been generated which are complementary to the ΔGAV°'s to obtain accurate rate coefficients without the need for imaginary frequencies or electronic energies of activation. These correlations depend on the temperature and activation energy of the exothermic step. The group additivity model has been successfully applied to a test set of reactions also calculated at the CBS-QB3 level of theory. A mean absolute deviation of 1.18 to 1.71 has been achieved showing a good overall accuracy of the model.
- This article is part of the themed collection: Bunsentagung 2018: Kinetics in the Real World
 Please wait while we load your content...
Please wait while we load your content...

