
Energy vs. density on paths toward more exact density functionals†

Abstract
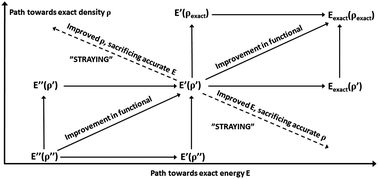
Recently, the progression toward more exact density functional theory has been questioned, implying a need for more formal ways to systematically measure progress, i.e. a “path”. Here I use the Hohenberg–Kohn theorems and the definition of normality by Burke et al. to define a path toward exactness and “straying” from the “path” by separating errors in ρ and E[ρ]. A consistent path toward exactness involves minimizing both errors. Second, a suitably diverse test set of trial densities ρ′ can be used to estimate the significance of errors in ρ without knowing the exact densities which are often inaccessible. To illustrate this, the systems previously studied by Medvedev et al., the first ionization energies of atoms with Z = 1 to 10, the ionization energy of water, and the bond dissociation energies of five diatomic molecules were investigated using CCSD(T)/aug-cc-pV5Z as benchmark at chemical accuracy. Four functionals of distinct designs was used: B3LYP, PBE, M06, and S-VWN. For atomic cations regardless of charge and compactness up to Z = 10, the energy effects of the different ρ are <4 kJ mol−1 (chemical accuracy) defined here as “normal”, even though these four functionals ranked very differently in the previous test. Thus, the “off-path” behavior for such cations is energy-wise insignificant. An interesting oscillating behavior in the density sensitivity is observed vs. Z, explained by orbital occupation effects. Finally, it is shown that even large “normal” problems such as the Co–C bond energy of cobalamins can use simpler (e.g. PBE) trial densities to drastically speed up computation by loss of a few kJ mol−1 in accuracy. The proposed method of using a test set of trial densities to estimate the sensitivity and significance of density errors of functionals may be useful for testing and designing new balanced functionals with more systematic improvement of densities and energies.
 Please wait while we load your content...
Please wait while we load your content...

