
Ab initio kinetics of the HOSO2 + 3O2 → SO3 + HO2 reaction†

Abstract
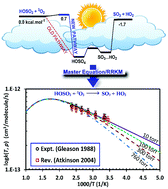
The detailed kinetic mechanism of the HOSO2 + 3O2 reaction, which plays a pivotal role in the atmospheric oxidation of SO2, was investigated using accurate electronic structure calculations and novel statistical thermodynamic/kinetic models. Explored using the accurate composite method W1U, the detailed potential energy surface (PES) revealed that the addition of O2 to a HOSO2 radical to form the adduct (HOSO4) proceeds via a transition state with a slightly positive barrier (i.e., 0.7 kcal mol−1 at 0 K). Such a finding compromises a long-term hypothesis about this channel of being a barrierless process. Moreover, the overall reaction was found to be slightly exothermic by 1.7 kcal mol−1 at 0 K, which is in good agreement with recent studies. On the newly-constructed PES, the temperature- and pressure-dependent behaviors of the title reaction were characterized in a wide range of conditions (T = 200–1000 K & P = 10–760 Torr) using the integrated deterministic and stochastic master equation/Rice–Ramsperger–Kassel–Marcus (ME/RRKM) rate model in which corrections for hindered internal rotation (HIR) and tunneling treatments were included. The calculated numbers were found to be in excellent agreement with literature data. The sensitivity analyses on the derived rate coefficients with respect to the ab initio input parameters (i.e., barrier height and energy transfer) were also performed to further understand the kinetic behaviors of the title reaction. The detailed kinetic mechanism, consisting of thermodynamic and kinetic data (in NASA polynomial and modified Arrhenius formats, respectively), was also provided at different T & P for further use in the modeling/simulation of any related systems.
 Please wait while we load your content...
Please wait while we load your content...

