
A molecular dynamics study of the complete binding process of meropenem to New Delhi metallo-β-lactamase 1†

Abstract
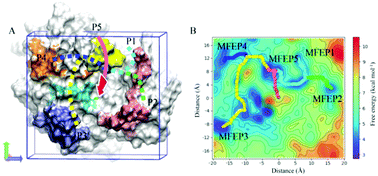
The mechanism of substrate hydrolysis of New Delhi metallo-β-lactamase 1 (NDM-1) has been reported, but the process in which NDM-1 captures and transports the substrate into its active center remains unknown. In this study, we investigated the process of the substrate entry into the NDM-1 activity center through long unguided molecular dynamics simulations using meropenem as the substrate. A total of 550 individual simulations were performed, each of which for 200 ns, and 110 of them showed enzyme–substrate binding events. The results reveal three categories of relatively persistent and noteworthy enzyme–substrate binding configurations, which we call configurations A, B, and C. We performed binding free energy calculations of the enzyme–substrate complexes of different configurations using the molecular mechanics Poisson–Boltzmann surface area method. The role of each residue of the active site in binding the substrate was investigated using energy decomposition analysis. The simulated trajectories provide a continuous atomic-level view of the entire binding process, revealing potentially valuable regions where the enzyme and the substrate interact persistently and five possible pathways of the substrate entering into the active center, which were validated using well-tempered metadynamics. These findings provide important insights into the binding mechanism of meropenem to NDM-1, which may provide new prospects for the design of novel metallo-β-lactamase inhibitors and enzyme-resistant antibiotics.
 Please wait while we load your content...
Please wait while we load your content...

