
Theoretical study of non-Hammett vs. Hammett behaviour in the thermolysis and photolysis of arylchlorodiazirines†

Abstract
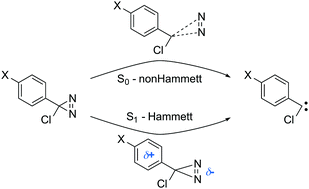
Arylchlorodiazirines (ACDA) are thermal and photochemical precursors of carbenes that form these molecules via nitrogen elimination. We have studied this reaction with multireference quantum chemical methods (CASSCF and CASPT2) for a series of ACDA derivatives with different substitution at the aromatic ring. The calculations explain the different reactivity trends found in the ground and excited state, with good correlation between the calculated barriers and the experimental reaction rates. The ground state mechanism can be described as a reverse cycloaddition with small charge transfer from the aromatic ring to the diazirine moiety. This is consistent with the lack of correlation between the Hammett σ descriptors and the experimental rates. In contrast, the excited state reaction is the cleavage of a single C–N bond mediated by small barriers of 4–6 kcal mol−1. The reaction path goes through a conical intersection with the ground state, which facilitates radiationless decay and explains the disappearance of the transient absorption signal measured experimentally. This leads to a diazomethane intermediate that ultimately yields the carbene. Electronically, excitation to S1 is characterized initially by significant charge transfer from the phenyl ring to the diazirine. The charge transfer is reversed during the C–N cleavage reaction, and this explains the preferential stabilization of the excited-state minimum by polar solvents and electron-donating substituents. Therefore, our calculations reproduce and explain the relationship found experimentally between the Hammett σ+ parameters and the life time of S1 (Y. L. Zhang, et al. J. Am. Chem. Soc., 2009, 131, 16652–16653).
 Please wait while we load your content...
Please wait while we load your content...

