
Excitation spectra of retinal by multiconfiguration pair-density functional theory†

Abstract
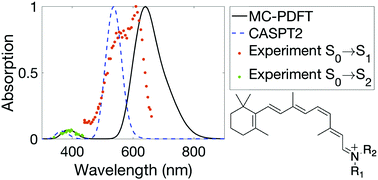
Retinal is the chromophore in proteins responsible for vision. The absorption maximum of retinal is sensitive to mutations of the protein. However, it is not easy to predict the absorption spectrum of retinal accurately, and questions remain even after intensive investigation. Retinal poses a challenge for Kohn–Sham density functional theory (KS-DFT) because of the charge transfer character in its excitations, and it poses a challenge for wave function theory because the large size of the molecule makes multiconfigurational perturbation theory methods expensive. In this study, we demonstrate that multiconfiguration pair-density functional theory (MC-PDFT) provides an efficient way to predict the vertical excitation energies of 11-Z retinal, and it reproduces the experimentally determined absorption band widths and peak positions better than complete active space second-order perturbation theory (CASPT2). The consistency between complete active space self-consistent field (CASSCF) and KS-DFT dipole moments is demonstrated to be a useful criterion in selecting the active space. We also found that the nature of the terminal groups and the conformations of retinal play a significant role in the absorption spectrum. By considering a thermal distribution of conformations, we predict an absorption spectrum of retinal that is consistent with the experimental gas-phase spectrum. The location of the absorption peak and the spectral broadening based on MC-PDFT calculations agree better with experiments than those of CASPT2.
 Please wait while we load your content...
Please wait while we load your content...

