
Ethanol synthesis from syngas over Cu(Pd)-doped Fe(100): a systematic theoretical investigation†
 *b
and
Gui-Chang
Wang
*ac
*b
and
Gui-Chang
Wang
*ac
Abstract
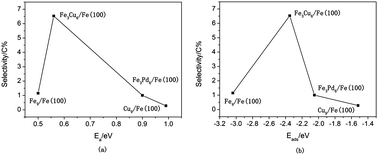
Although the reaction mechanism of syngas on Fe or Cu(Pd)-doped Fe has been studied extensively both experimentally and theoretically, the systematic prediction of the catalytic activity and selectivity for the formation of ethanol at the molecular level has not been reported to the best of our knowledge. In our present work, density functional theory calculations were performed to investigate the reaction mechanisms of the synthesis of ethanol, methanol, and methane from syngas over bimetallic Cu/Fe and Pd/Fe catalysts. Possible elementary steps involved in the formation of ethanol from syngas have been studied from thermodynamic and kinetic viewpoints. Our results show that an optimal route for the formation of ethanol on Cu/Fe and Pd/Fe catalysts starts with an initial process of the dissociation and hydrogenation of CO to produce CH3 species. Subsequently, the insertion of HCO groups into CH3 species leads to the formation of CH3CHO, followed by successive hydrogenation to form ethanol. The selectivity for ethanol is controlled by the formation of methyl species and the formation of C–C bonds between methyl species and CHO groups. Our kinetic model analysis shows that the selectivity for ethanol is highest on the Cu–Fe system, followed by Pd–Fe, and pure Fe (Cu) has the lowest selectivity. This is in close agreement with experimental findings in general. Possible reasons can be explained as follows: Fe sites favor the formation of CHx species, Cu and Pd sites are necessary to provide undissociated CO/HCO species, and Cu/Fe and Pd/Fe catalysts will provide dual active sites that are synergetic for chain propagation to generate precursors of C2 oxygenates by the insertion of CO/HCO groups into CHx species. The present results will further help the prediction of reaction activity and the design of efficient F–T catalysts for the formation of C2+ species to some extent.
 Please wait while we load your content...
Please wait while we load your content...

