
Molecular electrostatic potential on the proton-donating atom as a theoretical descriptor of excited state acidity†

Abstract
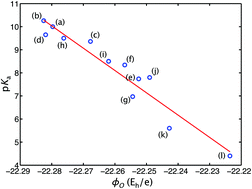
Organic photoacids with enhanced acidities in the excited states have received much attention both experimentally and theoretically because of their applications in nanotechnology and chemistry. In this study, we investigate the excited-state acidities of 14 hydroxyl-substituted aromatic photoacids, with a focus on using theoretical molecular electrostatic potential (MEP) as an effective descriptor for photoacidity. For these model photoacids, we applied time-dependent density functional theory (TDDFT) at the ωB97X-D/6-31G(d) level to calculate the molecular electrostatic potentials of S1 excited states and show that the molecular electrostatic potential on the proton-donating atom exhibits a linear relationship with the observed excited-state logarithmic acid dissociation constant (pKa*). As a result, the molecular electrostatic potential on the proton-donating atom can be used to estimate the pKa* values based on simple TDDFT calculations for a broad range of hydroxyl-substituted aromatic compounds. Furthermore, we explore the molecular electrostatic potential as a quantum descriptor for the photoacidities of cationic photoacids, and show a universal behavior of the pKa*–MEP dependence. We also investigate the solvent effects on the photoacidity using TDDFT calculations with implicit solvent models. Finally, we discuss the physical insights implicated by the molecular electrostatic potential as a successful measure for photoacidity on the mechanism of proton transfer in the molecular excited states. This pKa* descriptor provides an effective means to quantify the tendency of excited-state proton transfer with a relatively small computational cost, which is expected to be useful in the design of functional photoacids.
 Please wait while we load your content...
Please wait while we load your content...

