
Morphology control of 3-nitro-1,2,4-triazole-5-one (NTO) by molecular dynamics simulation†

Abstract
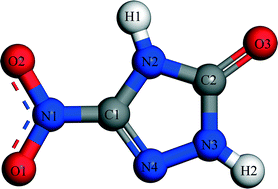
The vacuum, water-effect and ethanol-effect morphology of 3-nitro-1,2,4-triazole-5-one (NTO) is simulated using the attachment energy model by the molecular dynamics (MD) simulation method. The model size of the boundary layer dramatically influences the time and accuracy of the simulation. To study the crystal habit of NTO in a solvent within a short time and with high accuracy, the model size is first discussed. As the outcomes of NTO in water and ethanol are unsatisfactory, we propose the study of morphology control, which is performed by adding water to ethanol to adjust the growth rate of NTO surfaces. Based on the growth rate of each surface, we obtain a suitable mass ratio of ethanol/water, in which NTO is recrystallized and the crystal morphology is ameliorated. Besides, the radial distribution function (RDF) analysis shows that the solvent molecule adsorption on NTO molecules is mainly via solvent-crystal face interactions of hydrogen bonding. The simulation results can provide some guidance for the crystallization process of NTO, and the model size can also be applied in the morphology control of other compounds.
 Please wait while we load your content...
Please wait while we load your content...

