
Modelling uranyl chemistry in liquid ammonia from density functional theory†

Abstract
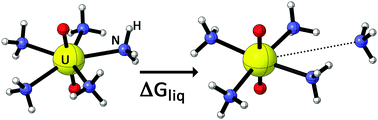
We developed a computationally efficient protocol based on Density Functional Theory (DFT) and a continuum solvation model (CSM) to predict reaction free energies of complexation reactions of uranyl in liquid ammonia. Several functionals have been tested against CCSD(T) and different CSMs have been assessed relative to Car–Parrinello Molecular Dynamics (CPMD) simulations in explicit solvent.
- This article is part of the themed collection: New molecules and materials from the f-block
 Please wait while we load your content...
Please wait while we load your content...

