
Towards predicting the power conversion efficiencies of organic solar cells from donor and acceptor molecule structures†
 ‡*a
Yongsheng
Chen
b
and
‡*a
Yongsheng
Chen
b
and
Abstract
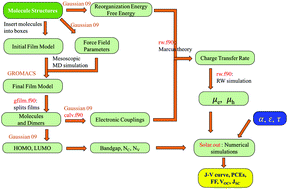
In this study, we developed a multiscale simulation framework to estimate the power conversion efficiencies of bulk heterojunction organic solar cells based on the molecular structures of the donor and acceptor. Firstly, we proposed a way to estimate the density of states (DOS) of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) in organic thin films based on quantum calculations, and verified the Gaussian-like DOS in the organic semiconductors. Secondly, the electronic couplings in these thin films were calculated. By adding PC71BM molecules, although the donor–donor couplings are not altered significantly, the charge mobility is enhanced via additional donor–acceptor and acceptor–acceptor couplings. Thirdly, random walk simulations were performed to estimate the charge carrier mobilities. Finally, by incorporating the calculated energy levels, mobilities and DOS of these bulk heterojunctions into the numerical model developed, we obtained the working curves and power conversion efficiencies, which are in general consistence with experiment results. This study builds the foundation for the computation of power conversion efficiencies of organic solar cells using fully atomistic simulations.
 Please wait while we load your content...
Please wait while we load your content...

