
Molecular simulation of the high temperature phase behaviour of α-unsubstituted sexithiophene

Abstract
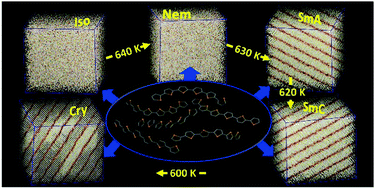
We examine the high-temperature phase behaviour of α-unsubstituted sexithiophene (α-6T) by means of Molecular Dynamics (MD) and Monte Carlo (MC) simulations using a recently developed state-of-the-art algorithm based on internal bridging moves. In the MD simulations, a realistic fully flexible united-atom model is used. In the MC simulations, a stiffer version of this united-atom model is implemented by restricting atoms on thiophene rings to remain strictly co-planar by employing holonomic constraints for all bond lengths and intra-ring bond bending angles; on the other hand, inter-ring torsion and bond bending angles are considered to be fully flexible subject to suitable potential energy functions. The MD simulations, which are started from the isotropic (Iso) phase at a relatively high temperature (above 700 K) and continued to lower temperatures under isobaric conditions using a very large simulation cell containing 8960 α-6T molecules, show four phase transitions: an isotropic-to-nematic (Iso-to-Nem) at 640 K, a nematic-to-smectic A (Nem-to-SmA) at 630 K, a smectic A-to-smectic C (SmA-to-SmC) at 620 K demonstrating smectic polymorphism, and a SmC-to-crystal-like (SmC-to-Cry) at 600 K. In the corresponding MC simulations, no Nem phase is observed; the system, as it is isobarically cooled down to lower temperatures from its Iso phase, undergoes directly a transition to a SmC phase at 690 K. This is attributed to the stiffer nature of the forcefield employed in these simulations. Both methods (MD and MC) shed light on the type and degree of molecular self-assembly, orientational and positional ordering as a function of temperature, and manifestation of liquid crystalline behaviour of α-6T. We provide a thorough characterization of structural ordering in all mesophases observed, in terms of several measures (radial correlation functions, orientational order parameters and X-ray diffraction patterns). According to the results, at the phase transition temperatures, drastic configurational changes take place driving α-6T molecules to positionally-ordered phases accompanied by self-assembly into characteristic layers which, in turn, are self-organized into macroscopic smectic phases. Our methodology opens up the way to exploring the rich phase behaviour and anisotropic ordering of the condensed phases of several longer (and perhaps more complex) thiophene-based polymers.
 Please wait while we load your content...
Please wait while we load your content...

