
Structure and dynamics of stereo-regular poly(methyl-methacrylate) melts through atomistic molecular dynamics simulations†

Abstract
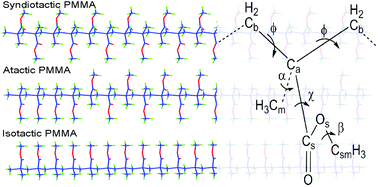
Poly(methyl-methacrylate), PMMA, is a disubstituted vinyl polymer whose properties depend significantly on its tacticity. Here we present a detailed study of the structure, conformation, and dynamics of syndiotactic, atactic, and isotactic PMMA melts at various temperatures (580, 550, 520, and 490 K) via all-atom molecular dynamics simulations. The calculated volumetric properties are close to experimental data. The Tg and chain dimensions of PMMA model systems are found to depend strongly on tacticity in agreement with experimental findings. The backbone bonds in trans (t), diads in tt, and inter-diads in t|t torsional states are the most populated for all PMMA stereo-chemistries and their fractions increase with the number of syndiotactic sequences. Also, the effective torsional barrier heights for the backbone, ester side group, and α-methyl group are larger for syndiotactic PMMA compared to the isotactic one. The structure of the PMMA chains is studied by computing the intra- and inter-chain static structure factors, S(q), and the radial pair distribution functions. In the first peak of S(q), both intra- and inter-chain components contribute, whereas the second and third peaks mainly come from inter- and intra-chain parts, respectively. For all PMMA stereo-isomers a clear tendency of ester-methyl groups to aggregate is observed. The local dynamics are studied by analyzing torsional autocorrelation functions for various dihedral angles. A wide spectrum of correlation times and different activation energies are observed for the motions of different parts of PMMA chains. The stereo-chemistry affects the backbone, ester side group, and α-methyl motions, whereas the ester-methyl rotation remains unaffected. The dynamic heterogeneity of the PMMA chains is also studied in detail for the different stereo-chemistries via the temperature dependence of the stretching exponent. Furthermore, the reorientational dynamics at the chain level and translational dynamics for monomer and chain centers-of-mass are analyzed.
 Please wait while we load your content...
Please wait while we load your content...

