
Machine learning meets volcano plots: computational discovery of cross-coupling catalysts†

Abstract
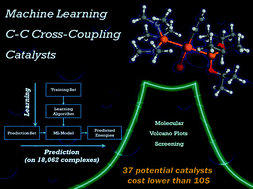
The application of modern machine learning to challenges in atomistic simulation is gaining attraction. We present new machine learning models that can predict the energy of the oxidative addition process between a transition metal complex and a substrate for C–C cross-coupling reactions. In turn, this quantity can be used as a descriptor to estimate the activity of homogeneous catalysts using molecular volcano plots. The versatility of this approach is illustrated for vast libraries of organometallic catalysts based on Pt, Pd, Ni, Cu, Ag, and Au combined with 91 ligands. Out-of-sample machine learning predictions were made on a total of 18 062 compounds leading to 557 catalyst candidates falling into the ideal thermodynamic window. This number was further refined by searching for candidates with an estimated price lower than 10 US$ per mmol. The 37 catalyst finalists are dominated by palladium phosphine ligand combinations but also include the earth abundant transition metal (Cu) with less common ligands. Our results indicate that modern statistical learning techniques can be applied to the computational discovery of readily available and promising catalyst candidates.
- This article is part of the themed collection: Most popular 2018-2019 physical and theoretical chemistry articles
 Please wait while we load your content...
Please wait while we load your content...

