
A combined experimental and computational study on the reaction of fluoroarenes with Mg–Mg, Mg–Zn, Mg–Al and Al–Zn bonds†

Abstract
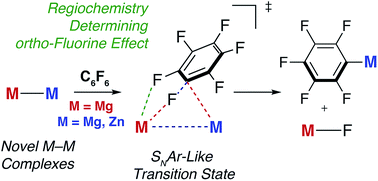
Through a combined experimental and computational (DFT) approach, the reaction mechanism of the addition of fluoroarenes to Mg–Mg bonds has been determined as a concerted SNAr-like pathway in which one Mg centre acts as a nucleophile and the other an electrophile. The experimentally determined Gibbs activation energy for the addition of C6F6 to a Mg–Mg bond of a molecular complex, ΔG‡298 K(experiment) = 21.3 kcal mol−1 is modelled by DFT with the ωB97X functional, ΔG‡298 K(DFT) = 25.7 kcal mol−1. The transition state for C–F activation involves a polarisation of the Mg–Mg bond and significant negative charge localisation on the fluoroarene moiety. This transition state is augmented by stabilising closed-shell Mg⋯Fortho interactions that, in combination with the known trends in C–F and C–M bond strengths in fluoroarenes, provide an explanation for the experimentally determined preference for C–F bond activation to occur at sites flanked by ortho-fluorine atoms. The effect of modification of both the ligand coordination sphere and the nature and polarity of the M–M bond (M = Mg, Zn, Al) on C–F activation has been investigated. A series of highly novel β-diketiminate stabilised complexes containing Zn–Mg, Zn–Zn–Zn, Zn–Al and Mg–Al bonds has been prepared, including the first crystallographic characterisation of a Mg–Al bond. Reactions of these new M–M containing complexes with perfluoroarenes were conducted and modelled by DFT. C–F bond activation is dictated by the steric accessibility, and not the polarity, of the M–M bond. The more open coordination complexes lead to enhanced Mg⋯Fortho interactions which in turn lower the energy of the transition states for C–F bond activation.
- This article is part of the themed collection: Most popular 2018-2019 main group, inorganic and organometallic chemistry articles
 Please wait while we load your content...
Please wait while we load your content...

